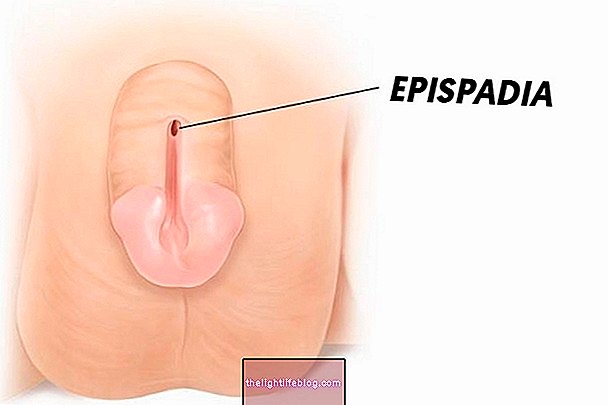

エディターズチョイス
ほとんどの訪問


テトラアメリア症候群は非常にまれな遺伝病であり、腕や脚を持たずに生まれ、骨格、顔面、頭部、心臓、肺、神経系または生殖器領域に他の奇形を引き起こす可能性があります。 この遺伝的変化は妊娠中であっても診断することができるため、特定された奇形の重症度に応じて産科医が出産後の生命を危険にさらす可能性があるので、 治療法はありませんが、四肢の欠如や軽度の奇形でしか生まれない場合があり、これらの場合には十分な生活の質を維持することができます。 Nick VujicicはTetra-amelia症候群で生まれました 主な症状 足と腕がないことに加えて、テトラアメリア症候群は、身体の様々な部分に多くの他の奇形を引き起こす可能性があります: 頭蓋骨と顔 カタラタス; 非常に小さい目; 非常に低いか、または欠けている耳。 鼻が非常に外れているか不在である。 口唇口蓋または口唇口唇。 心臓と肺 減少した肺の大きさ; ダイヤフラムが変化する。 心室は分離されていない。 心臓の片側の減少。 性器および尿路 腎臓の不在; 卵巣はよく発達していない。 肛門、尿道または膣の不在; 陰茎の下の穴の存在; 低生殖器官。 スケルトン 脊椎の不在; 小型または欠如した股関節の骨; 肋骨の欠如。 いずれの場合も、提示された奇形は異なり、したがって、平均寿命と生涯のリスクは子供によって異なる。 しかし、同じ家族内で罹患した人々
腎血管性筋腫は稀で良性の腎腫瘍であり、腎臓を傷つけるほどの大きさになるまでは通常症状がない。 従って、それは癌ではないが、腎臓の機能を損なうか、または重度の出血を引き起こす可能性があるので、特に4cmより大きい場合、血管筋脂肪腫は重大な問題であり得る。 その特定の原因はまだ分かっていませんが、血管筋脂質腫は40歳以上の男性でより一般的であり、ほとんど常に腎臓検査で誤って同定されています。 症状が腎臓癌である可能性があることを理解する。 主な症状 ほとんどの場合、血管筋脂肪腫は何の症状も引き起こさない。 しかし、次のような標識がある場合があります。 腹の側方領域の痛み; 血液を伴う尿。 頻繁な尿路感染症; 血圧の上昇。 さらに、腎臓出血が腫瘍によって引き起こされる場合、症状はより頻繁である。 これらの症例では、血圧の急激な低下、非常に重度の腹痛、薄くて非常に薄い肌を感じるなどの症状がみられます。 症状を評価し診断を確定するために、腎臓超音波検査医は、腎臓超音波検査、コンピュータ断層撮影または動脈造影などの様々な検査を行うことができる。 治療はどのように行われますか? 多くの場合、特に腫瘍が4cm未満の場合には、腎症の年1回の評価でのみ治療を行い、血管筋腫が発症しているかどうかを確認します。 しかし、血管筋脂肪腫の大きさが4cmを超える場合や症状を引き起こす場合は、腫瘍や腎臓の患部を除去

幼児期に通常発生する親しみやすい地中海熱をより良くコントロールし、共存させるためには、治療法はありませんが、小児科医に相談することをお勧めします。病気の危機はまれです。 したがって、地中海性熱の疑いがある場合、子供が発熱、腹部、胸部、関節の痛みを抱えている場合は、特に病気の原因となっている突然変異を特定し、診断を確認するために血液検査を行うことが重要です家族の病気のケース。 症状を和らげ、苦痛を軽減し、正常な生活を確保するための治療は、できるだけ早く開始し、医師の処方を使用して自宅で行うことができます。 しかし、発作時に症状が悪化し、息切れや失神がある場合は、直ちに救急室に行くことをお勧めします。 治療はどのように行われますか? 身体が炎症を制御または停止できない遺伝性の炎症性疾患である地中海での家族熱の治療には、以下のような医薬品の使用が含まれます: コルヒチン: 細胞増殖を防ぎ、症状の強さを低下させるため、体の炎症を軽減しますが、手足の下痢、衰弱、うずきなどの副作用を引き起こすことがあります。 AnacinraやRilonaceptなどの 抗炎症薬は 、主にコルヒチンで症状をコントロールすることができない場合に使用されます。 このタイプの治療は、病気を治癒させるものではありませんが、症状を緩和し、発症を予防するのに役立ちます。したがって、医師の監督下での危機の間、生涯にわたっ

craniofacial dysostosisとも呼ばれるCrouzon症候群は、頭蓋骨縫合の早期閉鎖があり、様々な頭蓋および顔の変形をもたらす珍しい状態である。 これらの変形はまた、視力、聴覚、または呼吸などの他の身体系の変化を生み出し、生涯にわたって矯正手術を行う必要がある。 疑わしい場合、診断は妊娠中、生まれて、または生後1年でさえ行われる遺伝学的細胞検査によって行われるが、通常は変形がより顕著になる2歳でしか検出されない。 主な症状 Crouzon症候群に罹患した子供の特徴は、奇形の重症度に応じて、軽度から重度までの範囲であり、 頭蓋骨の変形は、頭が高くなり、首が平らになります。 目がはみ出し、正常よりも遠い、拡大した鼻、斜視、乾性結膜炎、瞳孔の大きさの違いなどの顔面の変化; 眼の迅速で反復的な動き; IQは正常よりも低い。 難聴。 学習の難しさ。 心臓奇形; 注意欠陥障害; 行動の変化; 鼠径部、頸部、および/または腕の下の茶色から黒色の点。 Crouzon症候群の原因は遺伝的ですが、両親の年齢が介入してこの症候群で生まれる可能性が高くなります。なぜなら、年齢が高ければ高いほど、遺伝的変形の可能性が高くなるからです。 この症候群と同様の症状を引き起こす可能性のあるもう一つの疾患は、アパート症候群です。 この遺伝病の詳細をご覧ください。 治療はどのように行われますか? Cro

ゴーシェ病は、肝臓、脾臓、または肺のような身体の様々な器官、ならびに骨または骨髄に寄生する酵素的欠損によって特徴付けられる稀な遺伝病である骨。 したがって、罹患部位および他の特徴に応じて、疾患は3つのタイプに分けることができる: ゴーシェ病1型 - 非神経障害:これは最も一般的な形態であり、成人と小児の両方に影響を与え、正しい薬物療法を受けて進行が遅く、正常な生命が可能です。 ゴーシェ病2型 - 急性神経障害の形態:乳児に影響を及ぼし、通常は5ヶ月齢まで診断され、深刻な疾患であり、2年以内に死亡する可能性がある。 ゴーシェ病のタイプ3 - 亜急性神経障害の形態:それは小児および青年に影響し、その診断は通常6または7年後に行われる。 フォーム2ほど重症ではありませんが、神経および肺の合併症のために20歳または30歳までに死亡する可能性があります。 病気の重症度により、適切な治療を開始し、生命を脅かす合併症を軽減するために、できるだけ早く診断を行うべきである。 主な症状 ゴーシェ病の症状は、疾患の種類や罹患部位によって異なる場合がありますが、最も一般的な症状は次のとおりです。 過度の疲れ。 遅延成長; 鼻出血; 骨の痛み; 自発的骨折; 肝臓および脾臓の拡大; 食道の静脈瘤; 腹痛。 骨粗鬆症や骨壊死などの骨疾患もあります。 そして、ほとんどの場合、これらの症状は同時に起こることはありま
シャルコー・マリー・トゥース病(Charcot-Marie-Tooth disease)は神経および変性疾患であり、身体の神経および関節に影響を及ぼし、手で物体を保持する歩行および衰弱を困難または不可能にする。 多くの場合、この病気にかかっている人は車椅子を使う必要がありますが、長年生きて知的能力を維持することができます。 治療には、生命のために薬物療法と理学療法が必要です。 それがどのように現れるか Charcot-Marie-Tooth病気を示唆する徴候および症状には以下が含まれる: 足の曲線のような足の変化は、爪の上に非常に鋭く、指は鋭い。 歩行困難な人もいれば、足首の捻挫や骨折の原因となるバランスの欠如による頻繁な落ち込みがあります。 他の人は歩くことができません。 手に震える。 手の動きを調整するのが難しく、書くのが難しく、ボタンの服や料理をするのが難しい。 弱さと頻繁な疲れ。 腰椎痛および脊柱側弯症も見られる。 萎縮した脚、腕、手、足の筋肉; 脚、腕、手および足の接触および温度差に対する感受性の低下。 身体の痛み、けいれん、うずき、しびれなどの苦情は日常的には一般的です。 最も一般的なのは、子供が正常に発達し、両親が何かを疑っていないことです。3歳頃まで、脚の弱さ、頻繁な落ち込み、物の落下、筋量の減少、上記の他の信号。 治療はどのように行われますか? シャルコー・マリー・
FOP、進行性骨化筋炎または石男性症候群としても知られている進行性骨化線維化異形成は、靭帯、腱または筋肉などの軟部組織がゆっくりと骨に変わる非常にまれな遺伝病です体の動きを妨げ、体のイメージに大きな変化を引き起こす。 ほとんどの場合、症状は小児期に現れるが、組織の骨への変換は成人期に続き、診断が行われる年齢が変わる可能性がある。 しかし、出生時に赤ちゃんはすでに足指や肋骨の奇形をしているため、小児科医がこの疾患を疑う可能性があります。 進行性骨化線維化異形成の治療法はありませんが、腫れや関節痛などの症状を緩和し、生活の質を向上させる治療法があるため、小児科医を常に同伴することが重要です。 主な症状 進行性骨化線維化異形成の最初の兆候は、通常、足の奇形および場合によっては親指の存在を伴って出生直後に現れる。 その他の症状は、通常、20歳まで表示されます。 体によって赤くなったバンプは、消えても骨は残したままです。 穿刺部位における骨の発達; 手、腕、脚または足を動かす段階的な困難。 手足の血液循環の問題。 加えて、患部に応じて、特に頻繁な呼吸器感染症が発生した場合、心臓または呼吸器系の問題の発生が一般的です。 進行性肥厚性線維化異形成は、通常、首と肩に最初に影響を与え、その後、背中、胴、および手足に進行する。 この疾患は時間の経過と共にいくつかの制限をもたらし、生活の質を劇的に低下させ
ニーマン - ピック病は、同じ家族内で遺伝し、脳、脾臓または肝臓などのいくつかの器官に脂質蓄積を引き起こす、非常にまれな遺伝的症候群の群からなる。 罹患臓器および症状に応じて、ニーマン - ピック病は3つの主要な群に分けることができる: タイプA :最も重度のタイプであり、通常、生後1〜4ヶ月で生存率が4〜5歳に低下する。 タイプB :より重症のA型であり、成人まで生存できる。 タイプC :は、通常、小児期に発生するが、どの年齢でも発症することができる最も頻繁なタイプである。 この病気の治癒はまだありませんが、子どものQOLを向上させるために治療できる症状があるかどうかを評価するために小児科医を定期的に訪問することが重要です。 主な症状 ニーマン・ピック病の症状は、疾患の種類や罹患した器官によって異なりますので、各タイプの最も一般的な徴候は次のとおりです。 タイプA 約3か6か月で腹の腫れ; 成長が困難で体重が増えます。 最初の12ヶ月までの正常な精神発達。その後悪化する。 反復感染を引き起こす呼吸器の問題。 タイプB B型の症状は、Niemann-Pick A型疾患の症状と非常によく似ていますが、一般的にそれほど重度ではなく、小児期や青年期の後期に現れることがあります。 タイプC 動きを調整するのが難しい。 腹の腫れ; 目を垂直に動かすことの難しさ; 筋力の低下; 肝臓や肺の問題;

副腎白質ジストロフィーは、副腎、神経系および精巣に到達するX染色体に関連するまれな遺伝病です。 この疾患は特に男性に影響を及ぼし、どの年齢でも発現する可能性がある。 副腎白質ジストロフィーは、神経系の白い部分に存在するミエリンを変化させます。 神経系は一種の電気回路として機能し、ミエリンはこの回路から神経細胞を隔離する機能を有する。 ミエリンに変化がある場合、伝導は適切に行われず、神経系は機能を失う。 副腎白質ジストロフィーの症状 副腎白質ジストロフィーの症状が次第に現れます。 個人は、副腎機能、発言と相互作用の能力を失い、視力障害のために眼鏡をかけなければならず、歩行が困難であり、カテーテルを通して給餌を開始し、多くの痙攣を起こし、短時間で個体が昏睡状態。 あなたの体は家電製品の助けを必要としているので、すべてが起こります。 期待寿命 副腎白質ジストロフィーは、赤ちゃんの生後1ヶ月目に現れることがあり、この場合、平均余命は5年です。 人生の4〜10年の間に現れた場合、その子供は約10年の平均余命を持ち、成人の生活の中でその病気が現れたとき、その個体は何十年も生きることができるが、神経系の悪化同じように進歩的です。 治療はどのように行われますか? 副腎白質ジストロフィーの治療は、副腎のホルモンとLorenzoの油を使って行われ、病気の進行を遅らせる。 理学療法と心理療法も示されていま

トリメチルアミン尿症とも呼ばれる貧しい魚臭気症候群は、汗、唾液、尿および膣分泌物などの身体分泌物中の魚に似た強い臭いを特徴とする。 頻繁な入浴、日中の服装の変更、強い香水の使用などの措置は、魚や卵のような食物の摂取を避け、ダイエットによって主に行われるため、常に匂いを改善する助けとはならない。 症状 この病気の唯一の症状は、主に汗や息を介して身体から吐き出された魚の臭いですが、尿や膣分泌物にも悪い匂いがあります。 この匂いは、子供が母乳育児をやめ、正常な状態で食べ始めたとき、特に月経中には思春期に悪化し、避妊薬を使用するとさらに悪化する可能性があります。 原因 魚臭い症候群は、主に女性に影響を与える希少な遺伝病であり、魚介類、肝臓、エンドウ豆、卵黄などの食品中に存在するトリメチルアミンを消化する化合物が体内に存在しないことによって引き起こされます。 悪い体臭を引き起こす可能性のある救済 魚臭は主に遺伝的変化によるものですが、タモキシフェン、ケトコナゾール、スリンダク、ベンジダミン、ロスバスタチンなどのトリメチルアミンの蓄積を引き起こす医薬品を摂取すると、健康な人もこの症状を呈することがあります。 さらに、月経中に、またはストレス、発熱、腸内細菌叢の増加、魚の過度の摂取または肝臓または腎臓の問題のために、症候群のない人もこの悪臭を覚えるかもしれません。 診断 魚臭い症候群の診断は、血液
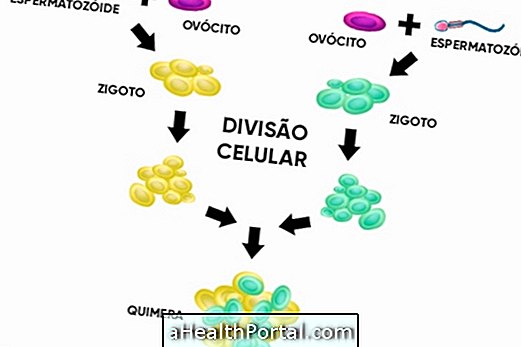
キメラリズムは非常にまれな遺伝的欠陥であり、ヒトが1種類以上のDNAを身につけている場合に発生します。 一般的に、この変化は症状を引き起こすことはありませんが、キャリアの身体に非対称性や変形などの変化や、めったにない場合には彼女の変態があるかもしれません。 このような状況は、遺伝物質の突然変異に起因する遺伝子改変を提示するモザイク現象とは異なる。 何が、そしてモザイクを特定するかを学ぶ。 キメラリズムは2つの方法で起こります。 天然または遺伝的キメラリズム それは、2つ以上の胚が1つに合併するときに起きるキメラ主義の古典的な形態である。 したがって、赤ちゃんは、次の図に示すように、2種類以上の異なる種類の遺伝物質で生まれます。 この変化の人は体内に2種類以上の異なるタイプのDNAを持つことがあり、DNAが同じ性別である場合、通常、人生に重大な影響はなく、身体の非対称性、例えば、肌の色や痛みの目、例えば。 しかし、もし彼らが異性の人であれば、その人は雌雄児に生まれます。 何が何であるかを理解し、自閉症を特定する方法。 2.人工キメラリズム 人が骨髄などの臓器移植を受け、その器官のDNAが人体の組織に取り込まれ、遺伝子検査によって検出されると起こります。 このような状況は、移植された人の体が臓器を良好に受け入れて、拒絶反応の危険性を減らすため、有益である。 キメラリズムの実証されているも

ウィーバー症候群は子供が小児期を通して非常に速く成長する稀な遺伝的状態であるが、知的発達の遅延を伴うだけでなく、大きな額や広い目のような特徴的な顔面特徴を有する。 場合によっては、関節や脊柱の変形、筋肉の弱い肌や弛緩した肌もあります。 ウィーバー症候群の治療法はありませんが、小児科医のフォローアップや症状の治療により、子供や両親の生活の質を向上させることができます。 主な症状 ウィーバー症候群の主な特徴の1つは、通常の成長よりも速いため、体重と身長は常に非常に高いパーセンタイルになっています。 しかし、他の症状や特徴には以下が含まれます: 低い筋力; 誇張された反射。 オブジェクトをつかむなどの自発的な動きの発達の遅れ; 私は低く泣き叫ぶ。 目は遠く離れている。 目の角に余分な肌; フラットネック。 長い額 非常に大きな耳。 足の変形; 手の指は常に閉じていた。 これらの症状のいくつかは、出生直後に特定することができ、他のものは、例えば、小児科医との相談の間、生後数ヶ月の間に同定される。 したがって、シンドロームは、出生後わずか数カ月で同定される場合がある。 さらに、症候のタイプおよび強度は、症候群の程度に応じて変化する可能性があり、したがって場合によっては気付かないこともある。 シンドロームの原因 ウィーバー症候群の発症の特定の原因はまだ分かっていないが、DNAコピーの一部を作るE

大田原症候群は、稀なタイプのてんかんであり、通常3ヵ月齢未満の乳児で起こるため、小児てんかん脳症としても知られている。 このタイプのてんかんの最初の発作は、通常、妊娠の最後の3ヵ月間に、まだ子宮内に起こりますが、赤ちゃんの生後最初の10日間にも起こることがあり、不随意の筋肉収縮が特徴であり、秒。 治療法はありませんが、発作の発症を予防し、子供の生活の質を向上させるために、医薬品の使用、理学療法、食物調節を行うことができます。 診断の確認方法 大田原症候群は、症状の観察と幼児の病歴の評価を通じてのみ、小児科医によって診断されることがある。 しかし、医師は脳波を発注することもあり、発作中の脳活動を評価する痛みのない試験です。 この試験の仕組みの詳細をご覧ください。 治療はどのように行われますか? 小児科医によって示される第1の治療法は、通常、クロナゼパムまたはトピラメートなどの抗杭薬を使用して発作の発症を制御することであるが、これらの医薬はほとんど結果を示さない可能性があるため、以下を含む治療の形態: コルチコステロイド またはプレドニゾンを 使用したコルチコステロイドの使用 :一部の子供の発作の数を減らす。 てんかんのための外科手術 :それは、発作が脳の特定の領域によって引き起こされ、脳の機能にとって重要でない限り、この領域の除去によって行われる小児に使用される。 ケトン生成食を取る
小人症はホルモンや医学的な問題の結果であり、身体を成長させずに発達させ、最大高さが1.47mになるようにし、矮星と呼ばれますが、低身長の人「身長」が最も受け入れられた表現である。 小人症には主に次の2種類があります。 比例または下垂体性小人症 : 体の すべての部分が正常より小さく、高さに比例して現れる。 不均衡または軟骨形成不全の神経 :身体の一部が予想以上の部分があり、身長の不均等感が生じます。 小人症には治癒はありませんが、子どもの発達に伴って起こる可能性のある合併症や正しい奇形の一部を治療で緩和することができます。 主な症状と原因 身長の低下に加えて、異なるタイプの小人症は、以下のような他の症状を引き起こすことがあります: 1.比例小人症 通常、このタイプの症状は生後1年目に現れます。その主な原因は、出生時に存在する成長ホルモンの産生の変化です。 症状としては、 3番目の小児科のパーセンタイル曲線以下の成長; 通常よりも少ない子どもの一般的な発達; 青年期の性的発達の遅延。 ほとんどの場合、診断は出生直後または小児相談中に小児科医によって行われます。 2.不規則な小人症 このタイプの小人症のほとんどの症例は、軟骨形成不全と呼ばれる軟骨の形成の変化によって引き起こされる。 これらの場合、主な症状と徴候は次のとおりです。 標準サイズのトランク。 短い脚と腕、特に前腕と大腿の領域で

Richieri-Costa Pereira症候群は、一部の赤ちゃんの17番染色体の遺伝的変化に起因する極めてまれな疾患であり、顔面および喉頭の異常や足および手の変形を引き起こす。 この病気は、USP病院で1992年にブラジルで最初に確認されました。この種の遺伝子変異を持つ子供はわずか20人しか同定されていません。 この症候群のほとんどの奇形は治癒しませんが、子どもの健康への影響を軽減し、生活の質を向上させるのに役立つ治療法があります。 主な症状および特徴 この病気の最も典型的な特徴は下顎骨の奇形であり、ちょうど骨ではなく、口の下の領域に裂け目を形成する2つに分かれています。 ただし、次のような他の特性も発生する可能性があります。 高さは通常よりも低い。 中央下部歯の欠如; 未開発の手; ベントまたは通常の指よりも小さい。 口が小さすぎる。 ベントフィート; 落ちた耳。 これらの変化のために、ほとんどの子供は、例えば、耳、喉頭または口の感染症を発症する傾向が強いだけでなく、呼吸、食べ、さらにはコミュニケーションが困難である。 シンドロームの原因 Richieri-Costa Pereira症候群は、17番染色体のDNAの変化によって引き起こされ、これらの変化につながる要因はまだ分かっていないが、研究者は20人の患児の約17人が同じ子孫。 このように、これは、特にこの家族につながってい

Hypermensiaは、非常に優れた自伝的記憶症候群とも呼ばれ、まれな症候群であり、キャリアは既にそれとともに生まれており、名前、日付、風景この症候を確認するためには、過去の出来事のいくつかの質問を含む認知と記憶のテストを行う必要があります。 このタイプの記憶を持つ人々は過去の出来事を思い出すことができ、記憶は非常に耐久性があり、シャープで活発です。 このまれな状態のキャリアは、脳内の記憶領域のより大きな発達を有する。 事象を覚えておく能力は認知の重要な領域であり、人々の間のよりよい推論と相互作用を可能にしますが、脳がより重要な事実に焦点を当てるためには、老いているか重要でない事実を忘れる能力も不可欠です。少ない摩耗。 主な特徴 高血圧の症状: 十分な生命と正確さで、新生児以降の事実を覚えておくこと。 記憶を強く不必要に保つ。 人生で一度しか見られなくても、日付、名前、番号を覚えやすく、景観や道を再現します。 したがって、この症候群の人は、過去または現在の事実を覚えている能力が高まり、数年前から完全に事実を覚えていて、通常は過去について考える時間を多く費やすことができます。 さらに、この症候群のほとんどの患者はこの状況にうまく対処できますが、それはあまりにも疲れてコントロールできないと考えている人もいます。 確認方法 Hypermnesiaは非常にまれな症候群であり、神経学者と心理
骨粗しょう症は、花崗岩骨疾患としても知られ、過剰な骨の成長を引き起こすまれな遺伝子変異である。 この突然変異は、年を経て密度が低下するのではなく、骨がより厚くて緻密になり、花崗岩よりも強くなります。 従って、強膜症は、骨粗鬆症のような骨疾患の発症を予防するが、未治療のまま放置すると、頭蓋骨内の圧力が増大して生命を脅かすなどの他の変化を引き起こす。 主な症状 硬化硬化症の主な徴候は骨密度の増加ですが、病気のことを知らせるいくつかの症状があります: 手の上に2, 3本の指を接合する。 鼻の大きさと厚さの変化; 頭蓋骨や顔の骨の誇張された成長; 顔の筋肉を動かすのが難しい。 指の先端が湾曲している。 指の爪の不在; 身長は平均以上です。 非常にまれな疾患であるため、その診断は複雑であり、したがって、医師は、すべての症状および臨床病歴を評価し、硬化の診断を示唆する前に、骨密度測定などの様々な検査を行う必要があります。 いくつかのケースでは、DNAと可能性のある変異を評価し、その疾患を引き起こすSOST遺伝子の変化を同定するのに役立つ遺伝子検査を命ずることができる。 それが起こる理由 硬化症の主な原因は、SOST遺伝子に起こる突然変異であり、スクレロスチン(骨密度を低下させ、生涯を通じて増加するタンパク質)の作用を減少させる。 通常、この病気は遺伝子のコピーが2つ変更された場合にのみ発生します
Berardinelli-Seipe症候群は、一般化した先天性脂肪異栄養症としても知られており、体内の脂肪細胞の機能不全を特徴とする稀な遺伝病であり、体内に脂肪が蓄積することはない肝臓や筋肉など)。 この症候群の主な特徴の1つは、通常8〜10年ぐらいの、思春期に始まる重度の糖尿病の発症であり、脂肪や糖分の少ない食事と糖尿病のコントロールを助ける薬で治療する必要があります。高コレステロール。 症状 Berardinelli-Seipe症候群の症状は体内の正常な脂肪組織の減少と関連しており、生後1年目に起こる可能性のある特徴をもたらす: 高コレステロールおよびトリグリセリド; インスリン抵抗性および糖尿病; 顎、大きい、細長い手と足; 筋肉の拡大; 肝臓や脾臓が増え、腹部に腫れが生じます。 心の問題。 加速した成長; 食欲の誇張された増加が、体重減少と; 不規則な月経周期; 厚くて乾燥した髪。 さらに、高血圧、卵巣の嚢胞および口の近くの首の側部の腫れなどの症状も現れることがある。 これらの症状は、小児期から観察され、思春期からより明らかになる。 診断と治療 この症候群の診断は、患者の臨床的特徴の評価およびコレステロール、肝臓、腎臓および糖尿病の問題を特定する試験に基づいて行われる。 診断の確認から、治療は主に糖尿病およびコレステロールを制御し、疾患の合併症を回避するためのものであり、メト

ハンハート症候群は、腕、脚または指の一部が完全に欠如することを特徴とする非常にまれな疾患であり、この状態は舌上で同時に起こり得る。 ハンハート症候群 の 原因 は遺伝的であるが、これらの遺伝子の変化につながる要因は説明されていない。 ハンハート症候群は治療法はありません が、整形手術は四肢の欠損を矯正するのに役立ちます。 ハンハート症候群写真 ハンハート症候群の症状 ハンハート症候群の主な症状は次のとおりです。 指または足の部分的または完全な欠如; 変形した腕と脚、部分的にまたは完全に欠けている。 小さいまたは変形された舌。 小さな口; 小さな顎。 顎を後退させた。 細かく変形した爪; 顔面麻痺; 嚥下困難; 睾丸の下降はない。 精神遅滞 一般的に、子供の発達は正常とみなされ、この病気の個体は、身体的な限界内で正常な生命を遂げることができる正常な知的発達を有する。 ハンハート症候群 の 診断 は、通常、妊娠中、超音波検査、および赤ちゃんが提示する徴候および症状の評価によって行われる。 ハンハート症候群の治療 Hanhart症候群の治療は、子供に存在する欠陥を修正し、生活の質を改善することを目指しています。 小児科医、整形外科医、整形外科医、理学療法士から、この症候群の罹患した各小児の症例を評価する一連の専門家の参加が必要です。 舌や口の欠陥に関連する問題は、咀嚼、嚥下および発語を改善
ウィリアムズ・ビューレン症候群は、心臓、協調、バランス、精神遅滞および精神運動の問題を提示するが、その主な特徴は子供の非常に友好的、超社会的およびコミュニケーション的行動である珍しい遺伝病である。 この症候群は、エラスチンの生成に影響を与え、血管、肺、腸および皮膚の弾性に影響を与える。 この症候群の子供は約18ヶ月で話し始めるが、韻や歌を学びやすく、一般的には音楽的感受性と聴覚記憶が多い。 彼らはしばしば、拍手、ミキサー、飛行機などの騒音を聞くと恐怖を覚えます。なぜなら、聴覚過敏と呼ばれる状態に過敏であるからです。 主な特徴 この症候群では、様々な遺伝子の欠失が起こる可能性があるため、ある個体の特徴は他の個体の特徴と大きく異なる可能性がある。 しかしながら、可能性のある特徴の中に存在し得る: 目の周りの腫れ 小さな険しい鼻 小さな顎 繊細な肌 青い目の人の星空の虹彩 出生時の僅かな長さと年間約1〜2cmの赤字 カーリーヘア 肉質の唇 音楽、歌、楽器の楽しみ 摂食の困難 腸の痙攣 睡眠障害 先天性心疾患 高血圧 反復耳感染症 視神経 非常に遠く離れた歯 頻繁な笑顔、コミュニケーションの容易さ 軽度から中等度までの知的障害 注意欠陥多動性障害 学校時代には、読解、スピーチ、数学の難しさ、 この症候群の患者は、特に思春期に、高血圧、耳炎、尿路感染症、腎不全、心内膜炎、歯の問題、ならびに脊柱







興味深い記事
推奨されます
ほとんどの訪問